

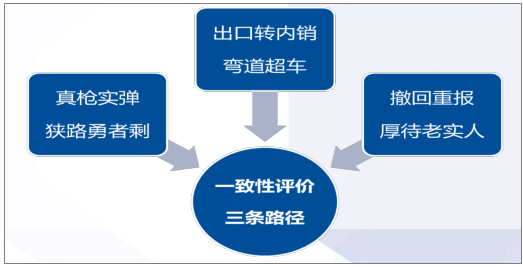
优质仿制药实现上市的三大路径逐渐清晰
与原研药质量/疗效一致是国际仿制药上市的基本前提,国内企业品种根据在品种竞争格局、研发基础、国际资源禀赋等方面的差异,选择不同的路径实现获批上市目标。
大多数的存量品种:客观上,中国目前大多数已上市的仿制药与原研药品在质量和疗效上存在不一致现象,需要弥补“历史旧账”。制药企业须积极进行品种梳理,开展药学研究,抢占国内BE试验资源,又快又好地完成仿制药一致性评价,抢占市场空间;
工业基础良好、国际化能力较强的企业的品种:在欧盟、美国、日本等发达市场上市的品种转向国内申报时,在优先审评及国外上市转国内申报审核后视同通过一致性评价的政策鼓励下,有望加快国内上市,实现弯道超车;
主动撤回重新申报企业品种:撤回后按与原研药一致性标准进行研究,重新申报将获得优先审评的政策倾斜,有望加快上市。
优质仿制药实现上市目标的三大路径
数据来源:公开资料整理
根据药品临床试验登记与信息公示平台的数据,通过试验方案和内容判断筛选后,截至2017年10月15日,预计属于一致性评价的BE共有135项。子公司合并后,这135项试验共涉及56家企业的72个仿制药品种。其中35项BE试验已完成。
一致性评价涉及仿制药品种繁多,任务量大
时间 | 药品批文总数(万) | 生产企业数(家) |
所有药品 | 18.9 | 4000左右 |
化学药品 | 12.2 | - |
仿制药品 | 11.6 | 3500-4000家 |
289基药品种 | 1.77 | 1859(含42家进口药品企业) |
数据来源:公开资料整理
已有30余个品种交CDE受理。通过跟踪数据发现,截至2017年10月29日,总局已统一受理18个改变处方工艺的一致性评价申请、8个未改变处方工艺或提出免于参加一致性评价的申请;此外,在总局统一受理前,共有8个左右的品种通过省局核查已提交至总局受理。这些品种有望成为第一批通过评价品种。
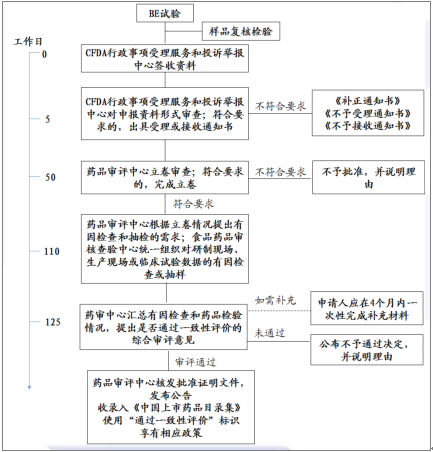
一致性申请审评审批耗时约6个月,2017年底至2018年初将有品种通过一致性评价。根据《总局关于仿制药质量和疗效一致性评价工作有关事项的公告(2017年第100号)》要求,一致性评价申请的审评工作一般应当在受理后的120个工作日内完成。我们预计2017年底至2018年初将有第一批仿制药品种通过一致性评价。
审评工作一般应当在受理后120个工作日内完成
数据来源:公开资料整理
CFDA规定三种视同通过一致性评价的情形,优待国内企业在国外上市的品种。CFDA针对“在欧盟、美国或日本批准上市”的药品规定了3种视同通过仿制药一致性评价的情况。其中,对于在欧盟、美国或日本批准上市的仿制药已在中国上市并采用同一生产线同一处方工艺生产的品种,申请人需提交境外上市申报的①生物等效性研究、②药学研究数据等技术资料,由CFDA审评通过后,视同通过一致性评价。其他情形或增加③处方工艺资料、④临床试验资料等。
部分企业选择出口转内销以实现弯道超车。由于国内BE临床试验机构资源相对紧张,完成仿制药一致性评价全过程时间较长(约20-28个月),部分国内优质企业选择在欧美日优先上市。这类企业生产水平较高,熟悉国际药品上市流程及国际市场药品销售渠道,在“出口转内销”上具备先发势,有望实现“弯道超车”。
对于未在国外上市的品种,通过优先审评鼓励企业走出去。除了已经获得海外ANDA批文的品种外,CFDA还对“申请人在美国、欧盟同步申请并获准开展药物临床试验的新药临床试验申请;在中国境内用同一生产线生产并在美国、欧盟药品审批机构同步申请上市且通过了其现场检查的药品注册申请”等国外未上市药物给予优先审评。截至9月25日,已有9家企业29项国内上市申请被纳入优先审评。这类药品将享受CFDA的审评资源倾斜,有望在国内快速上市并实现弯道超车。
相关报告:智研咨询发布的《2018-2024年中国仿制药行业深度调研及市场供需预测分析报告》
文章转载、引用说明:
智研咨询推崇信息资源共享,欢迎各大媒体和行研机构转载引用。但请遵守如下规则:
1.可全文转载,但不得恶意镜像。转载需注明来源(智研咨询)。
2.转载文章内容时不得进行删减或修改。图表和数据可以引用,但不能去除水印和数据来源。
如有违反以上规则,我们将保留追究法律责任的权力。
版权提示:
智研咨询倡导尊重与保护知识产权,对有明确来源的内容注明出处。如发现本站文章存在版权、稿酬或其它问题,烦请联系我们,我们将及时与您沟通处理。联系方式:gaojian@chyxx.com、010-60343812。
![2024年中国户用光伏行业现状及未来趋势分析:利好政策效应稳步释放,户用光伏并网容量持续增加[图]](http://img.chyxx.com/images/2022/0330/d1363a7ee3953fc25ed09e0b79158acce9dc7c22.png?x-oss-process=style/w320)
![2023年中国网络直播行业全景速览:用户体验持续优化,特色直播不断涌现[图]](http://img.chyxx.com/images/2022/0330/6b296592ed87ae76d174b4fbc262ff18a3c189b8.png?x-oss-process=style/w320)
![2024年中国风电制氢行业发展现状:行业技术不断提高,风电制氢有望实现大规模应用 [图]](http://img.chyxx.com/images/2022/0408/55d853aceb464ffcf6fad7c27bbd7795797b1b5a.png?x-oss-process=style/w320)
![2023年中国汽车冷冲压模具行业全景简析:新车型研发、上市加速,推动行业高速发展[图]](http://img.chyxx.com/images/2022/0330/ff5315f651f3e124d0f5a156ac51655e46e5433f.png?x-oss-process=style/w320)
![2024年中国钙钛矿电池行业发展现状分析:光伏企业加快布局钙钛矿,钙钛矿电池产业前景广阔[图]](http://img.chyxx.com/images/2022/0408/1ba88a0bac4b4a65439b806124f6fc0f4ab03cad.png?x-oss-process=style/w320)